
RNA-seq(遺伝子発現量 / トランスクリプトーム解析)
RNA配列を高い冗長度で網羅的に解析することにより、新規RNA検出に加えて、新規スプライス部位の検出や、定量発現解析が可能です。Total RNAサンプルからmRNAを精製し、cDNAを合成した後、次世代シーケンサーにより配列決定します。原核微生物、真核生物、参照配列の有無により、解析が異なります。遺伝子/トランスクリプトについて、サンプルごとに正規化発現量を求めます。反復数2以上の場合は、発現変動遺伝子を検出します。
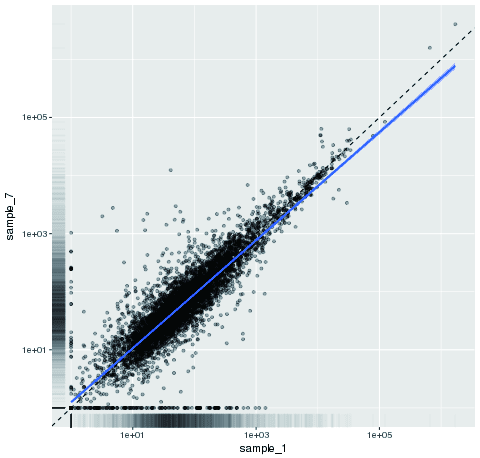
発現量解析
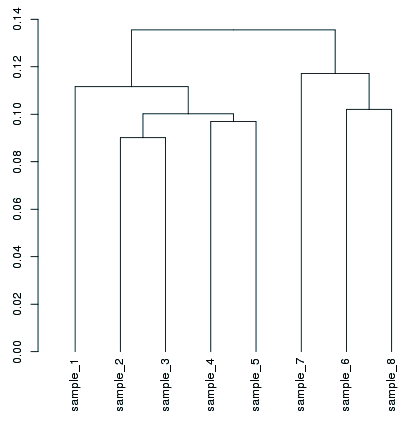
クラスタリング
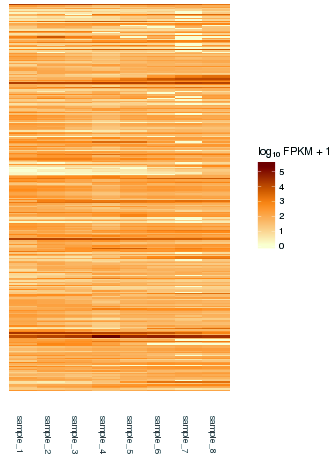
ヒートマップ
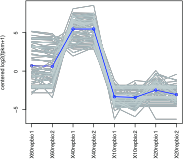
発現変動パターン
シーケンシング
Total RNAサンプルから、StandardまたはStrand-specific Libraryを作成します。
サンプルごとに異なるインデックス配列を付加することで、複数のサンプルを一度に解析することができ、1サンプル単位で安価に提供可能です。
| 細菌 | 真核 | 説明 | |
|---|---|---|---|
| mRNA精製法 (rRNA除去) | rRNA depletion | ・rRNA depletion ・polyA capture | rRNA depletionによるmRNA精製はnon-polyA RNAのシーケンスも可能にします。 |
| ライブラリー | ・Strand specific ・Standard | ・Strand specific ・Standard | Strand specificライブラリーでは転写産物の向きを保存したシーケンスが可能です。 一方、Standardライブラリーでは転写産物の向きに対するシーケンシングの向きはランダムです。 |
| リード長 | ・2x150bp〜 | ・2x150bp〜 | 既知の遺伝子構造に対して発現量を定量する場合は、1x50bpでも適切な情報が得られます。 一方、新規転写バリアント、融合遺伝子解析、de novoアセンブリーを行う場合には、2×150bpが推奨となります。 |
データ解析
リファレンスゲノムが存在する場合にはマッピング解析を行います。リファレンスゲノムが存在しない場合には、全サンプルのリード配列をアセンブリし、トランスクリプトーム配列を構築します。
- 発現量解析
トランスクリプトごとの正規化発現量を求め、サンプル間比較を行います。 - 発現変動遺伝子解析
反復数2以上の場合、正規化発現量を元にサンプル群で有意な発現変動を示す遺伝子/トランスクリプトを検出します。発現変動遺伝子について、階層的クラスタリングやGene Ontologyエンリッチメント解析等の高次解析も承ります。 - 新規発現領域、新規スプライス部位の検出(オプション)
- SNP解析や変異、融合遺伝子解析(オプション)
視覚化
マッピング結果や、SNP情報などをデータベース化・可視化することにより、発現量の比較以外にも様々な分析が可能です。

融合遺伝子解析の例
Iso-seq / cDNA-seq / Direct RNA-seq
RNAサンプルよりRT-PCRにより完全長cDNAを合成し、ライブラリ作成後、PacBioやNanoporeシーケンサーでシーケンスすることにより、完全長cDNA配列スプライシングバリアント等のアイソフォームを同定することが可能です。またNanoporeシーケンサーではRNA鎖を直接シーケンスするDirect RNA-Seqも実施可能です。
シーケンシング
mRNAからRT-PCRにより完全長cDNAを合成し、ライブラリ作成後、PacBioやNanoporeシーケンサーにより配列決定を行います。PacBioやNanoporeのロングリードを活かし、完全長cDNAを1本のリードとして配列決定することにより、アセンブルする必要が無く、スプライシングバリアント等のアイソフォームを連続配列として検出することが可能です。正確な転写開始位置の同定や融合遺伝子の探索も可能です。
データ解析
リファレンスゲノムへマッピングを行います。
- アイソフォーム解析
- アイソフォーム配列データを用いた、アノテーション解析

アイソフォーム解析の例