
動植物の品種改良に利用されるQTLマッピング、集団遺伝学、進化研究等においては、全ゲノム情報より少ない情報量でも有用な解析が可能である場合があります。
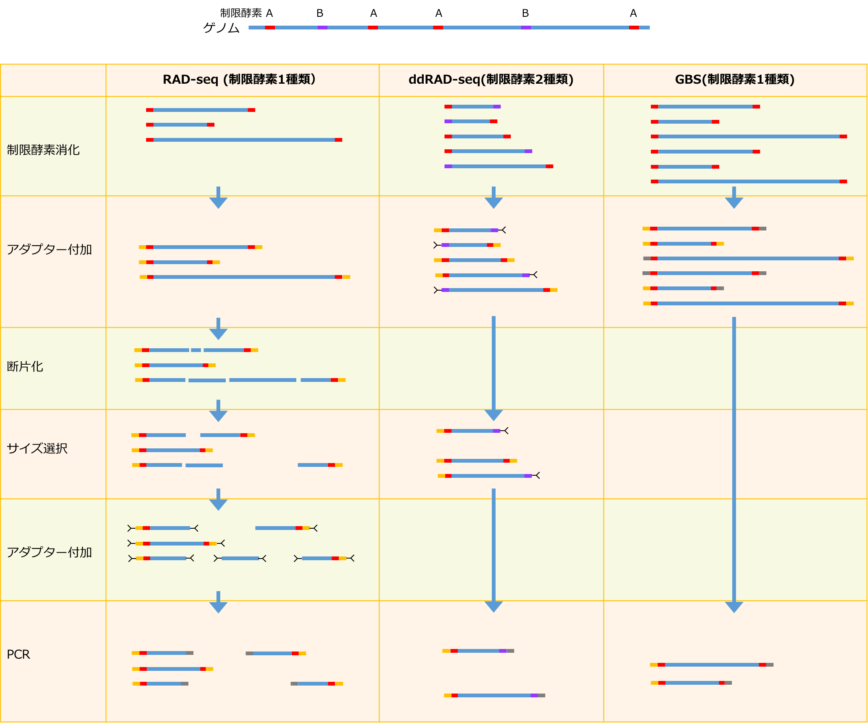
そこで、データ量を減らしながらも、サンプル間での比較を可能にする方法として、RAD-seq (Restriction Site Associated DNA Sequence)、ddRAD-seq(Double Digest RAD-seq)やGBS (Genotyping By Sequencing)等(下図)が開発されています。これらの方法では、ゲノムを制限酵素で消化し、切断サイトの近傍の配列を決定することで、その中に含まれるSNPを同定・比較します。本手法を用いることで、全ゲノムの0.1〜10%程度の配列を、ある間隔を持って決定することにより、解析に必要なシーケンシングデータ量を満たしながら、費用を抑えることが可能です。
本手法による配列決定部位の数は、ゲノムサイズ、制限酵素サイトの出現頻度、使用する制限酵素の数、ライブラリ作製方法に依存し、調節することが可能です。
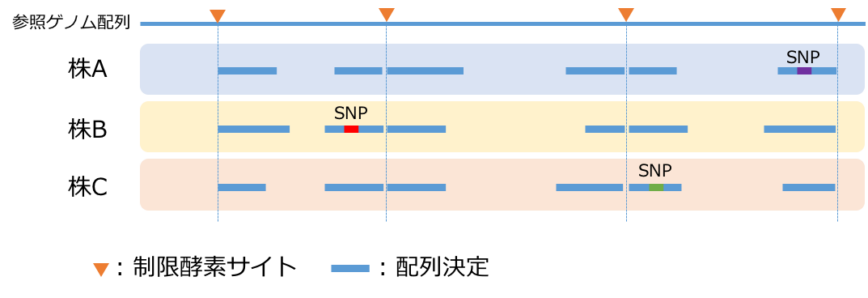
GBSによるジェノタイピング概念図
制限酵素を利用した配列決定のためのライブラリ作製方法
| RAD-seq | ddRAD-seq | GBS | |
|---|---|---|---|
| 1Mb中の座位数 | 30〜500 | 0.3〜200 | 5〜40 |
| データ量 | 中(他の2法よりも価格高め) | 少 | 少 |
| 特徴 | 断片化後サイズ選択するため、制限酵素サイトを含む断片が失われにくい | 2種類の制限酵素を使用し、RAD-seqよりもdepthが深い | ライブラリ構築ステップ、座位数が少ない |
参考文献:
Harnessing the power of RADseq for ecological and evolutionary genomics
Andrews, Kimberly R et al. 2016. Nat Rev Genet. 17(2): 81–92.